Congenital anorectal malformation is a digestive tract anomaly resulting from developmental disturbances of the hindgut during the embryonic period. It is the most common digestive tract defect in children, with an incidence rate of approximately 1 in 4,000 live births. Male infants are slightly more affected than female infants. Over 50% of congenital anorectal malformations are accompanied by fistula formation between the rectum and the urogenital system. Although the exact cause is unknown, it is believed to result from a combination of genetic and environmental factors.
Classification
The Wingspread Classification system for anorectal malformations was developed during the 1984 World Pediatric Surgery Meeting. This classification divides malformations into three categories based on the relationship between the rectal blind pouch and the levator ani muscle:
- High Malformation: The rectal blind pouch is located above the levator ani muscle.
- Intermediate Malformation: The rectal blind pouch is located within or slightly below the levator ani muscle.
- Low Malformation: The rectal blind pouch is located below the levator ani muscle.
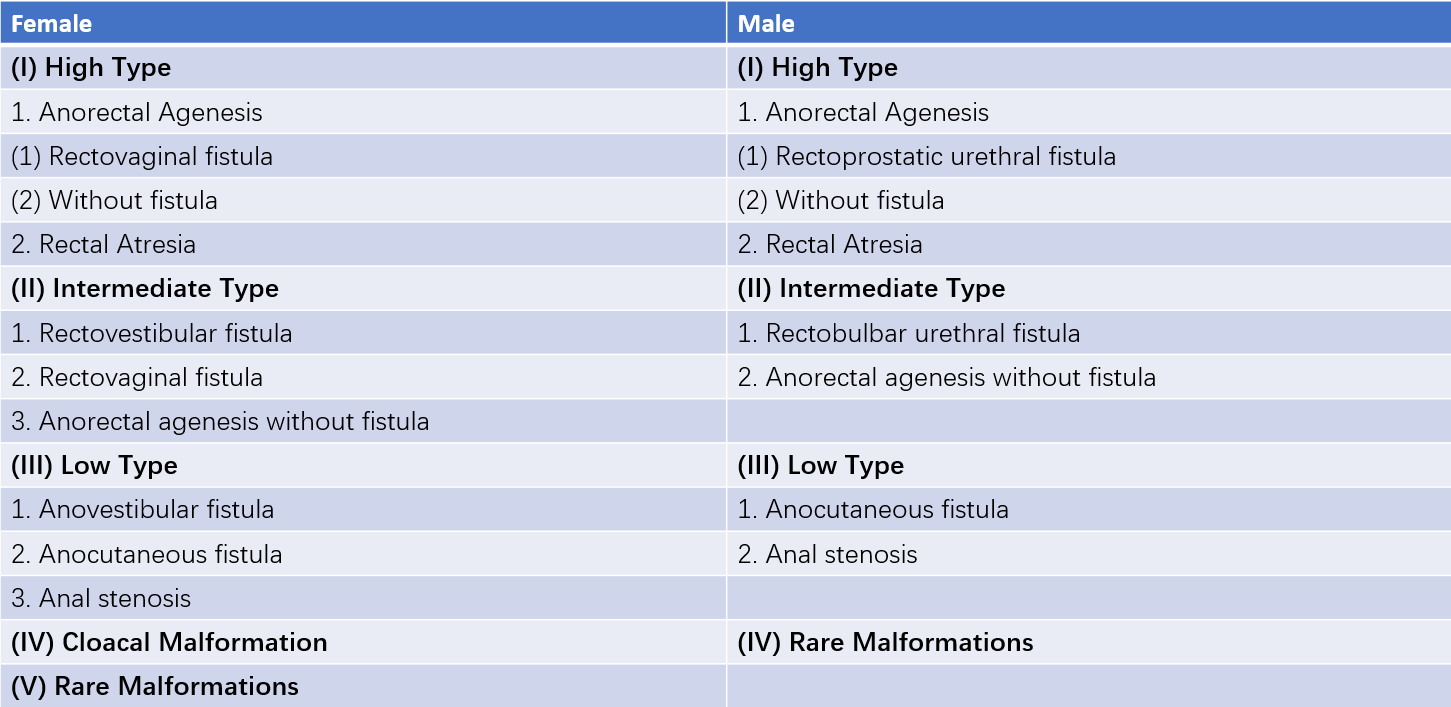
Table 1 Wingspread classification of anorectal malformations (1984)
Approximately 50% of anorectal malformations in boys are categorized as high malformation. Among girls, high malformation accounts for 20%, while low malformation accounts for about 40% in both sexes. The remaining cases are classified as intermediate malformation.

Figure 1 Congenital anorectal malformations (Without fistula group)
(1) Anal stenosis
(2) Low-type anal atresia
(3) High-type anorectal atresia
(4) Rectal atresia with normal anal development
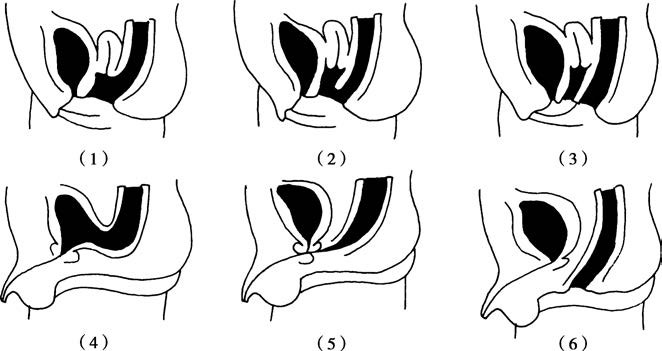
Figure 2 Congenital anorectal malformations (With fistula group)
Female:
(1) Rectovaginal fistula
(2) Rectovestibular fistula
(3) Rectoperineal fistula
Male:
(4) Rectovesical fistula
(5) Rectourethral fistula
(6) Rectoperineal fistula
In May 2005, during the International Meeting on the Diagnostic and Therapeutic Management of Anorectal Malformations held in Germany, a new classification system was proposed. It eliminates the high, intermediate, and low categories found in earlier systems. Instead, this classification is based on the type of fistula involved and includes rare types of malformations. This approach is designed to guide the selection of surgical techniques.
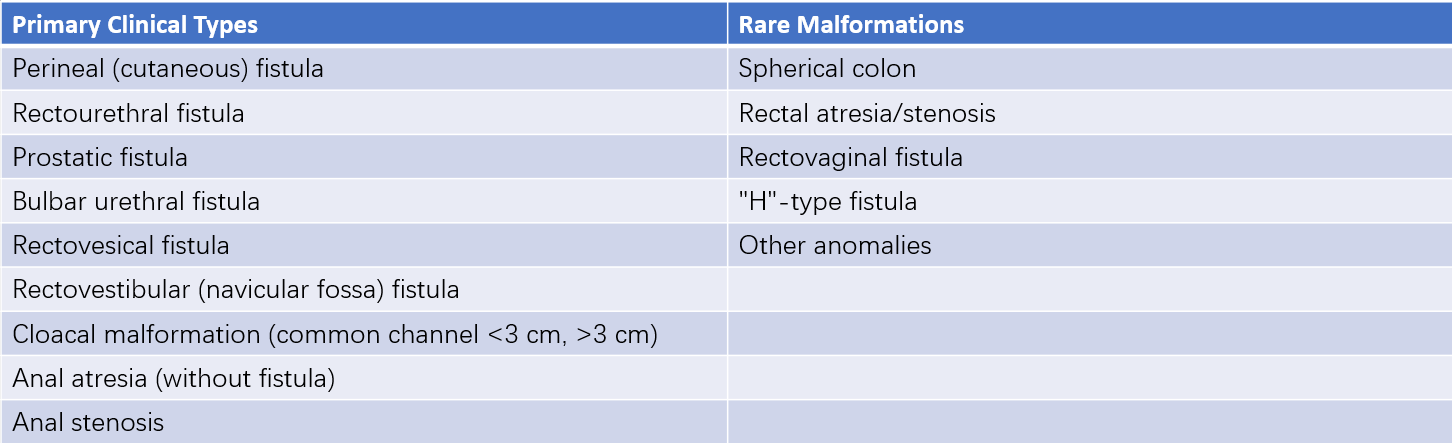
Table 2 International classification of anorectal malformations (2005)
According to this classification:
- Low Malformation: Includes perineal fistula, rectovestibular fistula, and anal stenosis.
- Intermediate Malformation: Includes urethral bulb fistula, anal atresia (without fistula), and most rectovaginal fistulas.
- High Malformation: Includes prostatic fistula and bladder neck fistula.
Clinical Manifestations
In the majority of cases, the absence of an anal opening at the normal anatomical location makes the condition easily detectable. Cases of anorectal malformation without fistula typically present with the absence of meconium passage within 24 hours of birth, accompanied by early vomiting and abdominal distension. When the fistula is narrow, preventing the discharge of meconium or allowing only small amounts to pass, symptoms include vomiting after feeding, followed by fecal-like vomiting and progressively worsening abdominal distension. In cases with a larger fistula, blockage symptoms may not appear initially, and difficulty in defecation may emerge weeks to years later.
For high rectal atresia with normal anal and anal canal anatomy, symptoms include failure to pass meconium or the discharge of murky liquid through the urethra. Digital rectal examination often reveals rectal atresia. Girls frequently present with rectovaginal fistula, while boys almost always exhibit rectal-urinary tract fistula. Passage of gas or meconium through the urethra is regarded as the primary symptom of rectal-urinary tract fistula.
Diagnosis
The diagnosis of congenital anorectal malformation is established when the absence of meconium passage (or only small amounts expelled through the urethra or perineal fistula) within 24 hours after birth is accompanied by vomiting and abdominal distension, which progressively worsen. Physical examination revealing no anal opening at the normal position confirms the diagnosis. Accurate determination of the level of rectal atresia, the presence and nature of fistulae, and associated anomalies remains critical. Additional examinations are performed to clarify the diagnosis and assess the extent of abnormalities:
Inverted X-ray Examination
Positioning of the rectal gas opacity aids in determining the location of the rectal blind pouch. The distance between the pelvic gas opacity and a metal marker represents the height of rectal atresia.
Urethral and Bladder Contrast Study, and Fistula Imaging
Visualization of contrast agents filling the fistula or entering the rectum provides valuable diagnostic information. For external fistulae, fistula imaging reveals the structure, length, and relationship between the fistula and the rectum.
Ultrasound Examination
Ultrasound provides a more accurate localization of the rectal blind pouch compared to X-rays. It demonstrates the distance between the rectal blind pouch and the anal skin, while observing the trajectory and length of the fistula.
Pelvic MRI or CT Scanning
Pelvic imaging reveals the developmental status of pelvic musculature and the position of the rectum. These imaging techniques offer clear visualization of the relationship between the rectal blind pouch and the muscular system, allowing precise determination of the extent and type of malformation. They are also useful for postoperative monitoring.
Treatment
The treatment strategy varies according to the type of anorectal malformation, but all cases require surgical intervention. Rectal atresia requires immediate surgery after birth.
Low Malformation
Surgery for low malformation is relatively straightforward and is typically performed via a perineal approach. For simple anal membrane atresia, treatment involves excision of the anal membrane followed by suturing of the rectal mucosa to the anal skin. For anal canal atresia, surgery involves mobilization of the rectal blind pouch, pulling it through the anal opening, and performing anal canal reconstruction.
High Malformation
High malformations require anorectal reconstruction through abdominal, perineal, or posterior sagittal incisions. Surgical principles include:
- Mobilization of the rectal blind pouch.
- Resection of associated fistulae and anatomical repair.
- Anorectal reconstruction.
Typically, a colostomy is performed first, followed by definitive surgery 6–12 months later.
In 2000, Georgeson pioneered laparoscopic anorectal reconstruction, which has since been widely and rapidly adopted for the treatment of high and intermediate anorectal malformations.