According to the WHO CNS 5 classification for neuro-oncology, diffuse gliomas are categorized into adult-type diffuse gliomas, pediatric-type diffuse low-grade gliomas, pediatric-type diffuse high-grade gliomas, circumscribed astrocytic gliomas, and ependymal tumors. The diagnosis of brain gliomas requires both histopathological findings and genetic characteristics. Key genetic markers include isocitrate dehydrogenase (IDH) mutation status, 1p/19q co-deletion status, O6-methylguanine-DNA methyltransferase (MGMT) promoter methylation, ATRX mutation, telomerase reverse transcriptase (TERT) promoter mutation, TP53 mutation, histone H3 mutation, MYB/MYBL1 gene alterations, MET gene alterations, and MAPK pathway gene mutations.
Diffuse gliomas have a high recurrence rate, and treatment following tumor recurrence remains a significant challenge in medicine. Reoperation is the leading treatment option, while patients unsuitable for surgery are recommended to participate in clinical trials.
Adult Diffuse Astrocytoma (IDH-Mutant, WHO Grade 2)
Adult diffuse astrocytoma primarily occurs in young to middle-aged adults, with a peak incidence between the ages of 25 and 45. These tumors are mostly located in the cerebral hemispheres, with the frontal and temporal lobes being the most common sites, followed by the parietal lobe and, less frequently, the occipital lobe. Astrocytomas exhibit slow growth, with an average disease history of two to three years, and progress gradually. Epilepsy is often the initial symptom; over 50% of patients present with seizures, and 75% experience headaches.
Diagnosis
Head CT scans typically reveal a hypodense intracranial lesion with relatively homogeneous features, minimal mass effect, and no significant peritumoral edema. On MRI, the tumor is often seen with long T1 and long T2 signals. Post-contrast enhancement scans generally show no enhancement, the tumor has poorly defined borders with brain parenchyma, and in rare cases, cystic components may be observed.
Treatment
Surgery is the primary treatment for low-grade astrocytomas, and early surgical intervention is commonly advocated. The goals of surgery include:
- Confirming the histological and molecular pathological diagnosis.
- Relieving mass effect and improving symptoms.
- Reducing tumor burden to delay progression.
- Preventing malignant transformation.
For patients with incomplete tumor resection or those over 40 years old, adjuvant radiotherapy is recommended postoperatively.
Adult Diffuse Astrocytomas (IDH-Mutant, WHO Grades 3–4) and Glioblastomas (IDH-Wildtype, WHO Grade 4)
These tumors, collectively referred to as high-grade gliomas, are more common in middle-aged and elderly individuals. The median age of onset is 45 years for grade 3 diffuse astrocytomas and 55 years for glioblastomas. High-grade gliomas exhibit rapid growth and a short disease course, with a median survival of 20 months for grade 3 gliomas and 16 months for grade 4 gliomas. Patients primarily present with symptoms of increased intracranial pressure and focal neurological symptoms, such as headaches, mental changes, limb weakness, and vomiting. Epileptic seizures occur less frequently.
Glioblastoma (GBM) represents the most aggressive astrocytic tumor. It can be classified into primary GBM and secondary GBM based on pathogenesis and clinical trajectory. Most cases are primary GBM, characterized by molecular genetic changes such as PTEN mutations and EGFR amplification and/or overexpression. Secondary GBM arises from the malignant transformation of grade 2 or 3 astrocytomas, with more than 90% of cases demonstrating a clinical history of a preceding low-grade tumor. These patients tend to be younger (average of 40 years), and key molecular genetic features include IDH mutations and TP53 mutations.
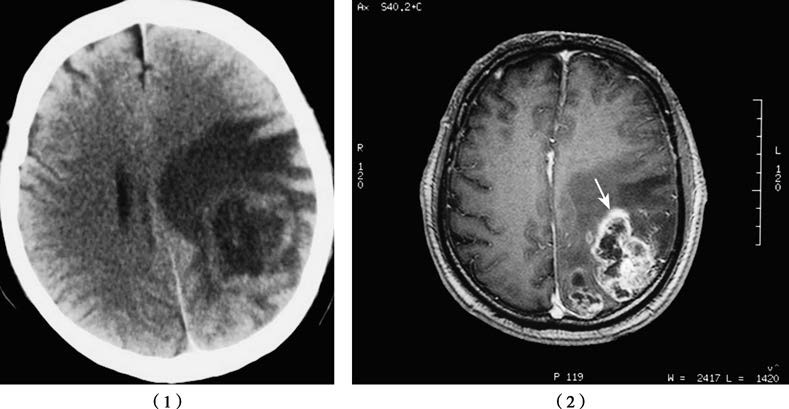
Figure 1 CT (1) and MR (2) images of a glioblastoma located in the left parieto-occipital region.
Diagnosis
In head CT scans, high-grade gliomas often appear as hypodense or inhomogeneous lesions with prominent mass effect and peritumoral edema. On MRI, 90%–95% of these tumors exhibit significant inhomogeneous enhancement, with features such as cystic degeneration, hemorrhage, and irregular tumor morphology.
Treatment
The treatment strategy for high-grade gliomas involves a multimodal approach, combining surgery with adjuvant radiotherapy and/or chemotherapy. The surgical resection principle is to remove as much tumor tissue as possible while preserving critical neurological functions. Surgical objectives include tumor debulking, alleviating mass effect, and establishing histological and molecular pathological diagnoses.
For newly diagnosed anaplastic gliomas, the standard treatment involves surgical resection followed by radiotherapy. Consideration for temozolomide chemotherapy is based on the tumor’s MGMT methylation status. The standard treatment for glioblastoma includes surgical resection, radiotherapy with concurrent temozolomide chemotherapy, and adjunctive tumor treating fields (TTFs).
Oligodendroglioma (WHO Grades 2–3)
Oligodendroglioma accounts for approximately 25%–33% of neuroepithelial tumors. According to the 2021 WHO classification, the definitive diagnosis of oligodendroglioma requires the simultaneous presence of IDH mutation and 1p/19q co-deletion. The peak onset occurs between the ages of 30 and 40, with a higher prevalence in males than females (3:2). The tumor grows relatively slowly, with an average course of about four years, and epilepsy is often the initial symptom (occurring in more than 80% of cases).
The most prominent radiographic feature of oligodendroglioma is calcification, visible on CT images in approximately 90% of cases. The tumor exhibits an infiltrative growth pattern, appears gray-red in color, and has a soft yet resilient consistency with relatively clear boundaries from normal brain tissue.
Given its sensitivity to chemotherapy, the recommended treatment approach involves combined surgical resection and chemotherapy. For anaplastic oligodendroglioma, radiotherapy may also be administered. Common chemotherapy regimens include:
- PCV protocol (Procarbazine + Lomustine [CCNU] + Vincristine).
- Monotherapy with Temozolomide.
Pediatric Diffuse Gliomas
Pediatric-type diffuse gliomas share some histological similarities with adult-type diffuse gliomas but differ significantly in terms of anatomical location and molecular pathological characteristics. While primarily found in children, these tumors may also occur in adults. The 5th edition of the WHO classification divides pediatric diffuse gliomas into two groups: pediatric-type diffuse low-grade gliomas and pediatric-type diffuse high-grade gliomas.
The diagnosis of pediatric-type diffuse gliomas is more complex compared to adult counterparts, due to differences in their histological features and molecular markers. Post-surgical radiation and chemotherapy for pediatric diffuse gliomas require cautious planning and administration.