Orbital rhabdomyosarcoma is the most common malignant orbital tumor in children, with most cases occurring in patients under the age of 8. It is rare in adolescents and occasionally observed in adults. The tumor exhibits rapid growth and a high degree of malignancy.
Clinical Manifestations
The tumor frequently originates in the superior orbit, causing the eye to protrude downward and forward. Swelling of the eyelids and conjunctiva may be significant, sometimes protruding beyond the palpebral fissure, resembling orbital cellulitis. Rapid tumor growth often leads to marked progression within days. A soft orbital mass may be palpable along the orbital rim. As the tumor grows rapidly, it may ulcerate through the conjunctival fornix, fixate the globe, cause vision loss, and involve the entire orbit with potential intracranial extension.
CT typically reveals a hyperdense soft-tissue mass within the orbit. Due to the rapid tumor growth, areas of necrosis may develop, resulting in inhomogeneous internal density. The tumor often appears irregularly shaped with poorly defined borders, along with evidence of bone destruction. Its invasive nature allows it to extend into surrounding structures. MRI typically shows the lesion as an intermediate signal intensity on T1-weighted imaging (T1WI) and high signal intensity on T2-weighted imaging (T2WI), with strong contrast enhancement. Areas of hemorrhage or necrosis may appear cystic in nature.
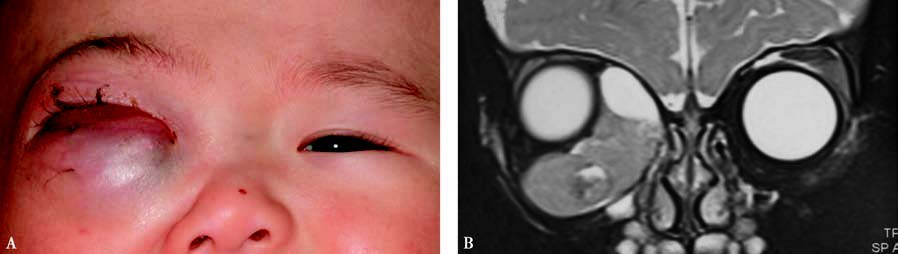
Figure 1 External and imaging features of right orbital rhabdomyosarcoma
A. Proptosis with conjunctival congestion and edema.
B. Coronal T2-weighted MRI showing a hyperintense signal with localized cystic changes.
Treatment
Current treatment strategies are based on clinical grouping, pathological type, TNM staging, and risk stratification. Multidisciplinary therapeutic approaches are often employed, including surgery, radiotherapy, and chemotherapy. For lesions that cannot be completely resected, a combination of radiotherapy and chemotherapy may be used prior to surgery.