Choroidal osteoma predominantly occurs in young women, often in a unilateral presentation. Its exact etiology remains unclear, although factors such as inflammation, trauma, hormones, and calcium metabolism may be involved. Patients are usually asymptomatic with good visual function, but secondary changes such as disruption of the retinal pigment epithelium, photoreceptor atrophy, subretinal fluid accumulation, and choroidal neovascularization can result in symptoms such as metamorphopsia, blurred vision, and visual field defects. The tumor is commonly located near the optic disc and appears as a yellow-white or orange-red flat elevation, often accompanied by pigment deposition. The lesion typically exhibits irregular borders resembling pseudopodia extending into the surrounding areas. Some patients may develop choroidal neovascularization, leading to subretinal hemorrhage or exudation. On CT imaging, choroidal osteoma is characterized by high-density signals similar to those of orbital bones, which is a classic diagnostic feature. Additional diagnostic tools such as fluorescein angiography (FFA), indocyanine green angiography (ICGA), and ocular ultrasonography can also aid in diagnosis.
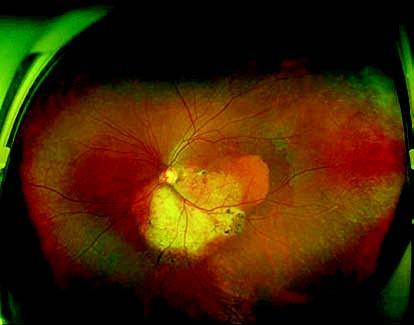
Figure 1 Wide-field fundus photograph of the left eye with choroidal osteoma
An oval yellow-white lesion is observed inferior to the optic disc in the temporal region, with well-defined margins measuring approximately 5 disc diameters (DD). The surface of the lesion contains neovascularization and scattered pigment deposits, with surrounding depigmentation of the retinal pigment epithelium.
Treatment
No definitive and effective treatment currently exists. Asymptomatic cases are primarily managed through clinical observation. For cases involving secondary subretinal neovascularization, anti-angiogenic therapy or laser photocoagulation may be used.