Ankylosing Spondylitis
Ankylosing spondylitis (AS) is an idiopathic inflammatory disease primarily affecting the axial skeleton, with an unclear etiology. Acute anterior uveitis occurs in 20-25% of patients with ankylosing spondylitis.
Clinical Features
The disease is common in young and middle-aged individuals, predominantly males. It is often associated with complaints of pain and stiffness in the lumbosacral region, which are usually more pronounced in the morning but improve with activity. Most patients present with acute, non-granulomatous anterior uveitis. Both eyes may be involved, though typically not simultaneously; instead, they are affected consecutively. The condition has a high recurrence rate, often alternating between the two eyes.
Diagnosis
The diagnosis is mainly based on clinical features such as lumbosacral pain, changes in the sacroiliac joint and spine, and the characteristics of uveitis. X-ray imaging may reveal changes including cartilage loss, bone erosion, sclerosis, joint space fibrosis, calcification, ossification, and bony ankylosis. Magnetic resonance imaging (MRI) or computed tomography (CT) can provide insight into early changes in the sacroiliac joint. The presence of the HLA-B27 antigen is also supportive of the diagnosis.
Treatment
Treatment of anterior uveitis includes topical corticosteroid eye drops and cycloplegic agents (details as outlined in the treatment of acute anterior uveitis). For systemic involvement, systemic corticosteroids and other immunosuppressants may be used. Consultation with a rheumatologist may be necessary in certain cases.
Vogt-Koyanagi-Harada Disease
Vogt-Koyanagi-Harada disease (VKH) is characterized by bilateral granulomatous panuveitis and is often accompanied by meningeal irritation, auditory disturbances, vitiligo, and depigmentation or loss of hair. Known as "idiopathic uveoencephalitis," it is one of the common forms of uveitis.
Etiology
The condition is caused by an autoimmune response, with genetic factors also implicated in its pathogenesis.
Clinical Features
Observations from a study of 410 cases of VKH revealed a typical clinical progression:
- Prodromal Stage (1-2 weeks before the onset of uveitis): Patients may experience nuchal rigidity, headache, tinnitus, hearing loss, and scalp hypersensitivity.
- Posterior Uveitis Stage (within 2 weeks of uveitis onset): Classic manifestations include bilateral diffuse choroiditis, optic disc inflammation, retinal neuroepithelial detachment, and retinal detachment.
- Anterior Uveitis Stage (2 weeks to 2 months after onset): In addition to posterior uveitis findings, signs of non-granulomatous anterior uveitis, such as small keratic precipitates (KP), anterior chamber flare, and anterior chamber cells, may appear.
- Recurrent Anterior Uveitis Stage (2 months after onset): Recurrent granulomatous anterior uveitis becomes prominent, often accompanied by sunset glow fundus changes, Dalen-Fuchs nodules, and ocular complications.
Not all patients exhibit all four stages. Timely treatment can halt the disease at a certain stage and may lead to complete recovery.
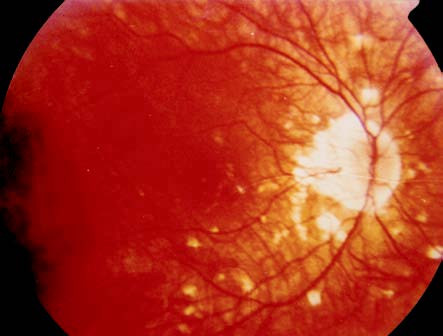
Figure 1 Fundus photography of a VKH disease patient
Depigmentation showing a sunset glow fundus and the presence of Dalen-Fuchs nodules.
Beyond the ocular findings mentioned above, extraocular features such as hair loss, depigmentation of hair, and vitiligo may occur at different stages of the disease. Common complications include complicated cataracts, secondary glaucoma, and exudative retinal detachments.
Diagnosis
Diagnostic criteria categorize VKH syndrome into early and late stages, both requiring the following three essential conditions to be met: no history of ocular trauma or intraocular surgery, bilateral eye involvement, and absence of significant infectious uveitis.
Early VKH Syndrome
Diagnosis can be established if any one of the following is present:
- Diffuse choroiditis or exudative retinal detachment.
- Serous retinal detachment with choroidal thickening.
- Multiple areas of early hyperfluorescence with late pooling on fundus fluorescein angiography (FFA).
- Persistent optic disc hyperfluorescence on FFA after corticosteroid treatment.
Late VKH Syndrome
Diagnosis can be established if recurrent bilateral granulomatous anterior uveitis is accompanied by any one of the following:
- Sunset glow fundus or migration of retinal pigment epithelium.
- Dalen-Fuchs nodules or multifocal chorioretinal atrophy.
- Window defects or moth-eaten hyperfluorescence on FFA.
For patients with severe media opacities, a clear history and features of early VKH syndrome must be present.
Treatment
For initial onset cases, oral prednisone is typically used, with an initial dosage of 0.6–0.8 mg/(kg·d). Dosage reduction usually begins 8–14 days later, with a maintenance dose of approximately 15–20 mg/day (adult dosage), and treatment often lasts for over a year. Recurrent cases are commonly treated with other immunosuppressive agents, such as chlorambucil, cyclophosphamide, cyclosporine, or azathioprine, often in combination with low-dose corticosteroids. For patients who do not respond to these medications, combined therapy with corticosteroids and anti-tumor necrosis factor (TNF) antibodies may be considered, though active infections, such as tuberculosis or hepatitis, as well as malignancies, should be ruled out prior to treatment. Regular follow-up is essential during systemic disease management. For secondary glaucoma and complicated cataracts, appropriate pharmacological or surgical interventions are required.
Behçet's Disease
Behçet’s disease is a multisystem disorder characterized by recurrent uveitis, oral ulcers, skin lesions, and genital ulcers. The condition is thought to be an autoinflammatory disease.
Etiology
The disease may be associated with bacterial or herpesvirus infections. Overactivation of Th17 and Th1 cells plays a critical role in its pathogenesis, and genetic factors also contribute to its development.
Clinical Manifestations
Ocular Involvement
The condition often manifests as recurrent non-granulomatous panuveitis, with hypopyon occurring in about 25% of cases. Typical fundus changes include retinitis and retinal vasculitis, with advanced stages frequently resulting in retinal vascular occlusion (so-called "ghost vessels"). Common complications include complicated cataracts, secondary glaucoma, proliferative vitreoretinopathy, retinal atrophy, and optic nerve atrophy.
Oral Ulcers
These are multiple, recurrent, and painful, typically lasting 7–14 days.
Skin Lesions
Lesions may present in varied forms, the most common being erythema nodosum, acneiform rashes, ulcerative dermatitis, and abscesses. Nodules or pustules at sites of needle punctures (positive skin hyperreactivity) represent a characteristic feature of Behçet’s disease.
Genital Ulcers
These are painful and may leave scars after healing.
Other Manifestations
Symptoms may include joint redness and swelling, thrombophlebitis, neurological involvement, gastrointestinal ulcers, epididymitis, and perianal abscesses.
Diagnosis
The diagnostic criteria proposed by the Japanese Behçet Disease Research Committee and the International Study Group for Behçet’s Disease are widely used.
The Japanese criteria classify patients into "complete" and "incomplete" types. The complete type is defined by the presence of four major criteria: recurrent uveitis, recurrent oral ulcers, polymorphic skin lesions, and genital ulcers. The incomplete type is diagnosed when three major criteria are present, or two major criteria along with additional findings.
The International Study Group’s diagnostic criteria require:
- Recurrent oral ulcers (at least three episodes within one year).
- Two of the following four features:
- Recurrent genital ulcers or genital scarring.
- Ocular involvement (anterior uveitis, posterior uveitis, vitreous cells, or retinal vasculitis).
- Skin lesions (erythema nodosum, pseudo-folliculitis, pustules, or acneiform nodules in post-adolescents).
- Positive skin hypersensitivity testing.
Treatment
Corticosteroids
Their long-term use at high doses is generally avoided. They are considered in the following situations:
- Anterior segment involvement, particularly in cases with hypopyon, may be managed with corticosteroid eye drops.
- Severe retinal inflammation or retinal vasculitis, which can rapidly damage vision, may require short-term high-dose administration.
- Combined use with other immunosuppressants, typically at a dosage of 20–30 mg/day.
Immunosuppressive Agents
Cyclosporine at 3–5 mg/(kg·d) is commonly used, with gradual dosage reduction after disease stabilization. Treatment duration generally exceeds one year. Other options include colchicine (0.5 mg, twice daily), azathioprine [1–2 mg/(kg·d)], chlorambucil [0.1 mg/(kg·d)], cyclophosphamide (50–100 mg/day), or mycophenolate mofetil (0.5–1 g/day). Regular monitoring of liver and kidney function, complete blood count, and blood glucose is required every two weeks during treatment. Dose reduction or discontinuation is advised if abnormalities are detected. Some medications may also cause infertility, necessitating regular semen analysis during treatment.
Various biologic agents, such as monoclonal antibodies targeting tumor necrosis factor (TNF) and interferon-α-2a, have been trialed for refractory Behçet’s disease. Possible infectious diseases should be ruled out before their use, and follow-up monitoring is recommended throughout therapy.
Cycloplegic Agents
These are employed in cases of anterior segment involvement.
Others
Complicated cataracts should be considered for surgical intervention only after complete control of inflammation. Secondary glaucoma may require pharmacological treatment, and surgical intervention should be considered if drug therapy fails.