Immunodeficiency diseases (ID) refer to a group of clinical syndromes caused by defects in immune cells (e.g., lymphocytes, phagocytes) or immune molecules (e.g., soluble factors such as interleukins, complement, immunoglobulins, and cell surface membrane molecules), resulting in impaired anti-infective immunity or immune dysregulation. Immunodeficiency diseases can be hereditary, caused by genetic mutations that impair immune system function, and are referred to as primary immunodeficiency diseases (PID). Immunodeficiency can also be acquired after birth due to environmental factors such as infections, nutritional imbalances, or certain diseases, and these are termed secondary immunodeficiency diseases (SID). Since SID is often milder, it is sometimes referred to as immunocompromised status. Immunodeficiency caused by human immunodeficiency virus (HIV) infection is specifically termed acquired immunodeficiency syndrome (AIDS).
Naming and Classification
In 1970, an expert committee of the World Health Organization (WHO) began categorizing PID. In 1999, this task was passed to the International Union of Immunological Societies (IUIS), which continues to update and classify these diseases approximately every two years. Diseases caused by genetic mutations leading to immune dysfunction exhibit diverse manifestations. The term "primary immunodeficiency disease," which has been widely used for several decades, emphasizes susceptibility to infections due to impaired anti-infective immunity but relatively overlooks other potential clinical manifestations of the disease. Since the 2017 classification of PID, the term "inborn errors of immunity" (IEI) has officially replaced PID to better reflect the spectrum of these disorders. In the latest 2022 IEI update and classification, a total of 485 IEI caused by genetic defects have been identified and categorized into 10 major groups: combined immunodeficiencies, combined immunodeficiencies with syndromic features, predominantly antibody deficiencies, immune dysregulation diseases, congenital defects of phagocytes, defects in intrinsic and innate immunity, autoinflammatory diseases, complement deficiencies, bone marrow failures, and IEI with phenocopies.
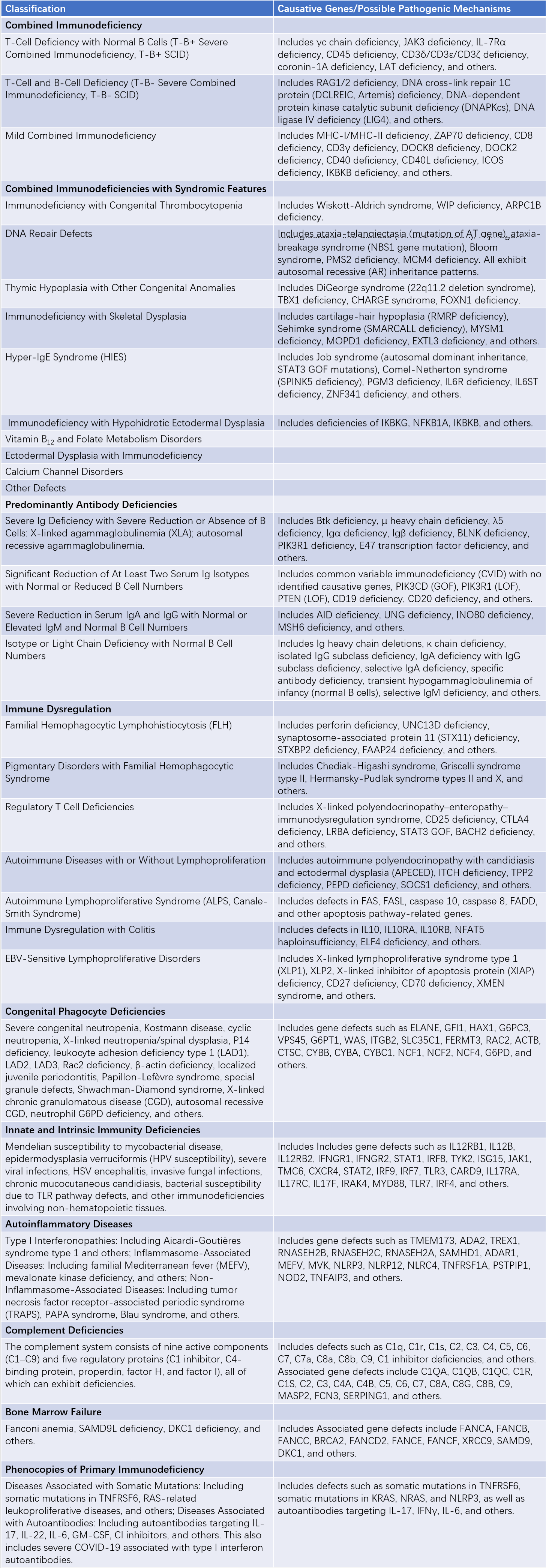
Table 1 Classification of congenital immunodeficiencies (IUIS 2022 version)
Common Types of PID
The widespread clinical application of immunological techniques, such as flow cytometry, and sequencing technologies, especially next-generation sequencing (NGS), has led to an increasing number of patients being diagnosed at the genetic or protein level. The primary types of PID confirmed genetically are concentrated in the following six diseases: X-linked agammaglobulinemia (XLA), X-linked hyperimmunoglobulin M syndrome (XHIM), Wiskott-Aldrich syndrome (WAS), X-linked chronic granulomatous disease (XCGD), X-linked severe combined immunodeficiency (XSCID), and common variable immunodeficiency (CVID).
X-linked Agammaglobulinemia (XLA)
Serum levels of IgM, IgG, and IgA are markedly reduced or absent, with decreased specific antibody levels. The number of immature B cells in the bone marrow is normal, but peripheral blood B cells are few or absent. Germinal centers in lymphoid organs are absent, and the number and function of T cells are normal. Mutations in the BTK (Bruton tyrosine kinase) gene in B cells are the cause. Disease onset typically occurs after 4 months of age, with a variable severity of symptoms. Patients are prone to recurrent pyogenic infections in the upper and lower respiratory tract and enterovirus infections. Untreated cases may result in life-threatening chronic lung disease.
X-linked Hyperimmunoglobulin M Syndrome (XHIM)
Circulating T cells are normal. IgM and IgD-positive B cells are present, but B cells expressing other types of Ig are absent. Serum levels of IgG, IgA, and IgE are generally reduced, while IgM levels may be normal or even markedly elevated. Clinical manifestations primarily include neutropenia, thrombocytopenia, and hemolytic anemia, often accompanied by biliary and liver disorders as well as opportunistic infections. Recurrent infections are characteristic, with severe respiratory infections typically occurring in early childhood.
Wiskott-Aldrich Syndrome (WAS)
This X-linked disorder commonly begins in infancy and is characterized by a triad of eczema, recurrent infections, and thrombocytopenia. Mild cases may present only with thrombocytopenia and small platelets, referred to as X-linked thrombocytopenia, and may sometimes be misdiagnosed as immune thrombocytopenic purpura. Immune function progressively declines, characterized by reduced IgM, poor antibody response to polysaccharide antigens, peripheral lymphopenia, and impaired cellular immunity. There is an elevated risk of lymphoma and autoimmune vasculitis. Mutations in the WASP (Wiskott-Aldrich syndrome protein) gene, located on the short arm of the X chromosome, are responsible for this condition.
Chronic Granulomatous Disease (CGD)
Mutations in the genes encoding components of the NADPH oxidase complex in phagocytes result in an inability to produce superoxide, singlet oxygen, and hydrogen peroxide (H2O2), leading to weakened pathogen-killing functions. This impairment results in chronic pyogenic infections and granuloma formation, particularly affecting lymph nodes, liver, lungs, and gastrointestinal tract. Pathogens often include Staphylococcus, Escherichia coli, Serratia, Nocardia, and Aspergillus. CGD can be X-linked, caused by mutations in the CYBB gene, which encodes the gp91phox protein, a component of the cytochrome b558 complex. It can also be autosomal recessive, caused by mutations in genes such as CYBA (encoding p22phox) or NCF1 and NCF2 (encoding p47phox and p67phox, respectively), all of which are proteins involved in electron transport.
Severe Combined Immunodeficiency (SCID)
T-Cell Defects with Normal B Cells (T-B+ SCID)
The most common cause is X-linked inheritance, due to mutations in the gene encoding the common gamma chain (γc) shared by receptors for IL-2, IL-4, IL-7, IL-9, and IL-15. Severe bacterial and viral infections typically occur shortly after birth, with most patients dying in infancy.
Absence of both T and B Cells (T-B- SCID)
This type is autosomal recessive.
- RAG1/2 deficiency: Mutations in the RAG1 or RAG2 genes result in a marked decrease in peripheral T and B cell counts, typically presenting in infancy.
- Adenosine deaminase (ADA) deficiency: Mutations in the ADA gene lead to the accumulation of toxic intermediate metabolic products, inhibiting T and B cell proliferation and differentiation. Most patients develop infections early in life, but mild cases may present in older children or adults.
- Reticular dysgenesis: A defect in the development and maturation of lymphoid stem cells and myeloid precursor cells causes severe reductions in peripheral lymphocytes, neutrophils, and platelets, often resulting in death during infancy.
Common Variable Immunodeficiency (CVID)
This group of syndromes is characterized by varying degrees of immunoglobulin deficiency with an unknown etiology and uncertain inheritance patterns. Clinical manifestations typically begin in older children or young adults and include recurrent respiratory infections such as sinusitis, pneumonia, and bronchiectasis, as well as gastrointestinal infections and enteroviral meningitis. Patients are prone to lymph node enlargement, splenomegaly, and a high incidence of lymphatic system malignancies, gastrointestinal cancers, and autoimmune diseases. Serum IgG and IgA levels are low, while IgM is normal or reduced. Diagnosis relies on excluding other primary immunodeficiency diseases. B cell counts may be reduced, but T cell dysfunction, such as altered CD4+/CD8+ ratios and reduced IL-2, IL-5, and IFN-γ activity, may play a key role in disease pathology.
Common Clinical Features
The clinical manifestations of PID vary greatly depending on the underlying cause, but common features include recurrent infections, increased susceptibility to tumors, and autoimmune diseases. A family history of the disorder may be apparent in some cases. It is worth noting that earlier attention was primarily directed toward infections in PID, but recent discoveries indicate that many PIDs are primarily characterized by immune dysregulation, including autoimmune reactions, allergic responses, and uncontrolled inflammatory responses, often with only mild or no infections.
Recurrent and Chronic Infections
Infections are the most common manifestations of immunodeficiency and are characterized by recurrent, severe, persistent, and difficult-to-treat infections. Unusual or low-virulence microorganisms are often the causative agents. Some children require continuous use of antibiotics to prevent infections.
- Age at onset of infections: Symptoms appear before 1 year of age in 40% of cases, between 1–5 years in 40%, between 6–16 years in 15%, and in adulthood in only 5% of cases. T cell and combined immunodeficiencies tend to present shortly after birth, while antibody deficiencies usually manifest later, between 6–12 months, after maternal antibodies are no longer present. Adult cases are most frequently associated with CVID.
- Infection sites: The respiratory tract is most commonly affected, leading to recurrent or chronic otitis media, sinusitis, conjunctivitis, bronchitis, or pneumonia. Gastrointestinal infections, such as chronic enteritis, are also prevalent. Skin infections may present as pustules, abscesses, or granulomas. Some cases involve systemic infections, including sepsis, meningitis, and bone or joint infections.
- Pathogens: Antibody deficiencies are commonly associated with pyogenic bacterial infections. T cell deficiencies predispose to intracellular pathogens such as viruses, Mycobacterium tuberculosis, and Salmonella species, as well as fungi and protozoa. Complement deficiencies often lead to Neisseria infections. Neutrophil dysfunction is frequently associated with Staphylococcus aureus. Infections are often caused by weakly virulent, opportunistic pathogens.
- Course of infections: Episodes are often recurrent or prolonged, with poor responses to treatment. Antimicrobial therapy, particularly bacteriostatic agents, may show limited efficacy, requiring bactericidal agents at higher doses and longer durations for adequate effect.
Non-immune factors, such as congenital airway abnormalities or foreign bodies in the airway, may also contribute to recurrent infections. These factors should be ruled out when considering a diagnosis of PID.
Autoimmune Diseases
Survivors who do not succumb to severe infections may develop autoimmune diseases as they age. Common autoimmune conditions associated with PID include hemolytic anemia, idiopathic thrombocytopenic purpura, neutropenia, systemic vasculitis, systemic lupus erythematosus, dermatomyositis, immune complex nephritis, type 1 diabetes, autoimmune hypothyroidism, and arthritis.
Tumors
There is a particularly high risk of lymphatic system tumors, with rates tens to hundreds of times greater than those in the general population. B cell lymphomas are most common, but lymphocytic leukemia, T cell lymphomas, Hodgkin's disease, adenocarcinomas, and other cancers may also occur.
Other Clinical Manifestations
In addition to the shared clinical features, other specific characteristics may aid in diagnosis. Examples include growth retardation or failure to thrive, disseminated or localized infections following Bacillus Calmette-Guérin (BCG) vaccination, eczema and bleeding tendencies in Wiskott-Aldrich syndrome, unique facial features, congenital heart defects, and difficult-to-control hypocalcemic seizures in thymic aplasia.
Diagnosis
Medical History and Physical Examination
Previous History
Delayed separation of the umbilical cord is an important clue for diagnosing type I leukocyte adhesion deficiency (LAD). Severe or prolonged cases of measles or chickenpox suggest cellular immune deficiencies. Information about factors causing secondary immunodeficiency diseases, previous history of blood transfusions, use of blood products, or occurrences of graft-versus-host reaction (GVHR) should be obtained. Vaccination history, particularly any occurrence of paralysis following administration of live poliovirus vaccines, should be documented in detail.
Family History
Approximately one-fourth of affected children have family members who died early in life from infections. Performing pedigree investigations of the child's family can be helpful. In some cases, the propositus of PID may represent the initial occurrence of a genetic mutation and lack a positive family history. Information about family members with allergic diseases, autoimmune disorders, or malignancies can aid in assessing the propositus.
Physical Examination
Severe or recurrent infections may result in weight loss, delayed development, malnutrition, mild-to-moderate anemia, and hepatosplenomegaly. Deficiencies in B cells may result in reduced or absent peripheral lymphoid tissue, such as the tonsils and lymph nodes. X-linked lymphoproliferative disease may present with generalized lymphadenopathy. Signs of infection, such as skin abscesses, stomatitis, periodontitis, or oral thrush, may be observed. Specific syndromes may exhibit additional distinctive features, such as thymic hypoplasia, symptoms of Wiskott-Aldrich syndrome (WAS), and ataxia-telangiectasia (AT).
Laboratory Tests
The diagnosis of PID relies on laboratory immunological assessments and genetic analyses. Repeated infections of unknown cause, early-onset autoimmune diseases, and a positive family history raise the suspicion of PID. Conclusive diagnosis requires laboratory evidence to determine the nature of the immune deficiency. Since it is not currently feasible to assess all immune functions, and certain experimental methods are only available at specialized research centers, laboratory evaluations are typically conducted in three stages:
- Initial screening tests;
- Further diagnostic evaluations;
- Specialized or research-based assays.
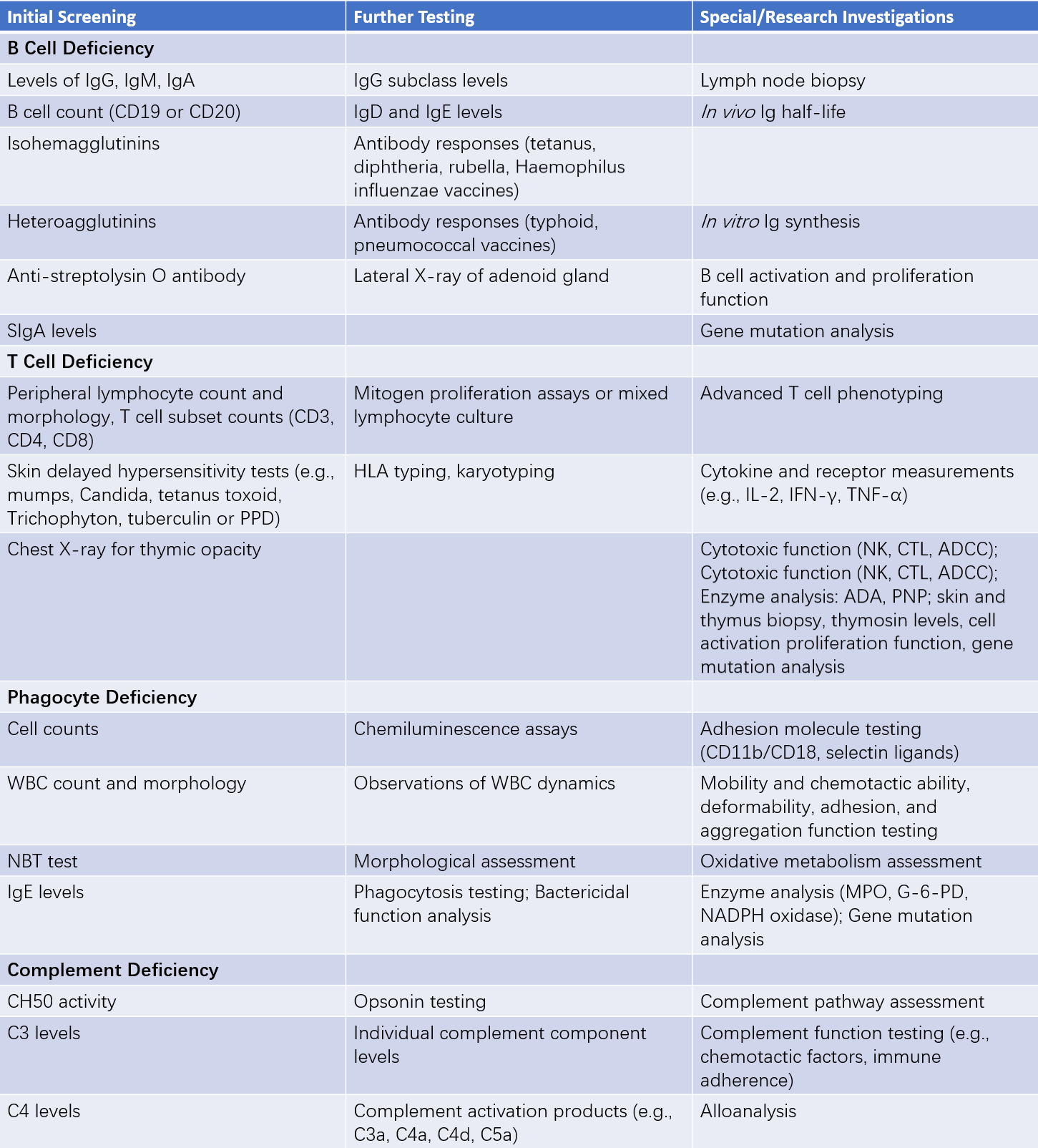
Table 2 Laboratory testing for immunodeficiency diseases
Note:
ADA: Adenosine deaminase
ADCC: Antibody-dependent cellular cytotoxicity
CTL: Cytotoxic T lymphocyte
G-6-PD: Glucose-6-phosphate dehydrogenase
MPO: Myeloperoxidase
NADPH: Nicotinamide adenine dinucleotide phosphate
NBT: Nitroblue tetrazolium test
NK: Natural killer cells
PNP: Purine nucleoside phosphorylase
Among these, initial screening tests play a particularly crucial role during early diagnostic processes.
Measurement of Immunoglobulins (Ig)
Serum levels of IgG, IgM, IgA, and IgE are assessed. In older children and adults, total Ig levels above 6 g/L are considered normal, while levels below 4 g/L or IgG levels below 2 g/L suggest antibody deficiencies. Total Ig levels between 4–6 g/L or IgG levels between 2–4 g/L are considered borderline, necessitating additional assessments such as antibody response tests or IgG subclass analysis. During early childhood (up to 2–3 years of age), Ig levels may fall below the normal range for age without clinical manifestations of recurrent infection, and immune deficiencies may not need to be considered unless symptoms arise. Elevated IgE levels may indicate abnormalities in phagocyte function, particularly in cases of chemotactic defects.
Anti-A and Anti-B Isohemagglutinin Titer
This test assesses IgM function. Normally, infants over six months of age have anti-A and anti-B isohemagglutinin titers of at least 1:8. In WAS patients with low IgM levels, isohemagglutinin titers are reduced or undetectable.
Antistreptolysin O (ASO) and Heterophile Agglutinin Titers
Due to exposure-induced production of natural antibodies, heterophile agglutinin titers are usually greater than 1:10 in the general population, reflecting IgG function. Where antimicrobial exposure is common, ASO titers are generally low. If ASO levels remain below 50 units after 12 years of age, this may suggest IgG antibody response deficiencies.
Secretory IgA (SIgA Levels)
SIgA deficiency is commonly associated with selective IgA deficiency. It is typically assessed in saliva, tears, nasal secretions, and gastric fluid.
Absolute Peripheral Blood Lymphocyte Count
Approximately 80% of peripheral blood lymphocytes are T cells, so absolute lymphocyte counts can reflect T-cell quantity. Normal values range from (2–6) × 109/L; levels below 2 × 109/L are suspicious for T-cell reduction. In infants, absolute lymphocyte counts below 3 × 109/L warrant further evaluation and reassessment. Persistent values below 3 × 109/L necessitate immune function testing to identify the cause. Counts below 1.5 × 109/L in infancy strongly suggest SCID.
Chest X-Ray
Absence of the thymic opacity in infants and young children indicates T-cell function impairment. However, the thymus may be obscured in the mediastinum, requiring careful interpretation.
Delayed Cutaneous Hypersensitivity (DCH) Test
This test evaluates Th1 cell function. Antigens are injected intradermally, and localized reactions are observed 24–72 hours later. Redness and induration indicate a positive result, suggesting normal Th1 function. Commonly used antigens include mumps vaccine, tuberculin purified protein derivative (PPD), Mucor extract, Candida extract, and diphtheria toxoid. Negative results may occur in children under 2 years of age due to lack of prior sensitization. Testing with five or more antigens is recommended; if at least one produces a positive result, Th1 function is considered normal.
Nitroblue Tetrazolium (NBT) Test
NBT is a pale yellow soluble dye that is reduced by activated neutrophils to form blue-black granules. Reduction rates exceeding 90% are considered normal. Reduction rates below 1% are indicative of chronic granulomatous disease. Carriers of the disease exhibit mosaic patterns.
Complement CH50 Activity and C3/C4 Levels
Normal total CH50 activity ranges from 50–100 U/mL. Normal C3 levels vary by age, ranging from 570–1950 mg/L, while normal C4 levels range from 70–400 mg/L, depending on the specific age group.
Genetic Mutation Analysis and Prenatal Diagnosis
Most PIDs are monogenic disorders. Sequence analysis of the disease-associated gene can identify mutations, aiding in confirming the diagnosis and conducting family studies. Genetic mutation analysis is also the best method for prenatal diagnosis, alongside other techniques such as measuring enzyme activity levels (e.g., ADA) in chorionic villus samples.
Treatment
General Management
Children with PID require specialized pediatric care, including prevention and treatment of infections, appropriate isolation measures, attention to nutrition, and enhanced family education to build confidence in coping with the disease. Post-treatment, children should receive encouragement to participate in normal activities to the extent possible. Infection sites must be promptly treated when identified, and in some cases, long-term prophylactic antimicrobial therapy may be necessary. Chronic lower respiratory tract infections, even in the absence of clinical symptoms, require regular chest imaging and pulmonary function monitoring.
For children with T-cell deficiencies, to prevent GVHR, blood or blood products must be irradiated at doses of 20–30 Gy before use. Blood donors should undergo CMV screening. Tonsillectomy and lymph node removal are generally discouraged, and splenectomy is contraindicated. If children retain some antibody production capacity, inactivated vaccines like the DTP vaccine can be administered. Live vaccines are contraindicated in patients with severe immunodeficiency to prevent vaccine-related infections. Families with confirmed cases of immunodeficiency should undergo genetic counseling, and prenatal screening should be conducted during pregnancy, with termination considered when necessary.
Replacement Therapy
Intravenous Immunoglobulin (IVIG)
This form of treatment is indicated for patients with low IgG levels. In some cases, symptoms completely resolve following IVIG therapy, allowing for normal growth and development. The recommended dosage is 100–600 mg/kg administered intravenously once per month, continued for life. Dosages are individualized based on infection control.
Special Immune Serum Globulins (SIG)
SIG, including preparations for varicella-zoster, rabies, tetanus, and hepatitis B, is used for prophylaxis in high-risk children.
Plasma
Plasma contains IgG, IgM, IgA, complement, and other immunologically active components. The usual dose is 20 mL/kg, with the possibility of increasing the dose if needed.
Other Replacement Therapies
Fresh Leukocytes
These are administered during severe infections in patients with phagocyte deficiencies. Fresh leukocytes are short-lived in the body, and repeated administration may trigger adverse immune reactions. For this reason, their use is reserved for severe infections rather than routine replacement therapy.
Cytokine Therapy
This includes treatments with agents such as thymic peptides, transfer factor, IFN-γ, and IL-2.
Enzyme Replacement Therapy
For adenosine deaminase (ADA) deficiency, infusions of red blood cells (rich in ADA) or intramuscular injections of bovine ADA-polyethylene glycol conjugates are performed, with the latter showing superior outcomes compared to red blood cell transfusions.
Immune Reconstitution
Immune reconstitution involves the implantation of normal cells or genetic fragments into the patient to correct immune deficiencies on a long-term basis.
Thymus Transplantation
This includes transplantation of thymic tissue or thymic epithelial cells. Successful thymus transplants have been reported, particularly for severe DiGeorge syndrome.
Hematopoietic Stem Cell Transplantation (HSCT)
HSCT is currently the primary curative treatment for PID globally. Reports of successful HSCT treatments for conditions such as SCID, XHIM, WAS, and CGD have shown success rates of 65%–85%. Matched sibling donors are the ideal donors, with success rates exceeding 90%.
Fetal Liver Transplantation
Some children have demonstrated successful engraftment following fetal liver transplants, but this method is rarely used today.
Bone Marrow Transplantation (BMT)
Over 1,000 children with PID have undergone BMT globally.
Umbilical Cord Blood Hematopoietic Stem Cell Transplantation
Umbilical cord blood, rich in hematopoietic stem cells, serves as an important source of cells for immune reconstitution. Compared to matched unrelated marrow donor (MUD) transplants, umbilical cord blood stem cell transplantation results in reduced GVHR severity.
Peripheral Blood Stem Cell Transplantation
Peripheral blood stem cells can also be used for transplantation.
Gene Therapy
The mutated genes responsible for many forms of PID have been identified, and their mutation sites have largely been determined, providing the foundation for gene therapy. Gene therapy involves integrating normal target gene fragments into the genome of the patient's stem cells (gene transduction). These genetically modified cells replicate through mitosis, ensuring the continual presence of the transformed gene within the patient.
Gene therapy for PID has been under investigation for many years, with over 10 clinical trials completed globally, achieving certain levels of success. It is expected to become a critical therapeutic tool in pediatric care over the next decade. Gene editing aims to repair mutated target genes in situ, potentially avoiding the risk of tumorigenesis associated with random chromosomal integration during gene transduction. This approach may represent an ideal method for future PID gene therapy. However, as of now, gene editing remains in the preclinical research phase.