Ewing sarcoma is a small round cell sarcoma exhibiting varying degrees of neuroectodermal differentiation. It is characterized by small round cells containing glycogen. The tumor primarily affects children and is often found in the diaphysis of long bones, the pelvis, and the scapula. In 85% of cases, a specific chromosomal translocation t(11;22)(q24;q12) can be identified. This translocation fuses the 5′ end of the EWSR1 gene on chromosome 22q12 with the 3′ end of the FLI1 gene, a member of the ETS family on chromosome 11q24, forming the oncogenic fusion gene EWSR1::FLI1.
Clinical Features
The primary symptoms include localized pain and swelling, which progressively worsen over time. Systemic conditions may deteriorate rapidly, often accompanied by low-grade fever, leukocytosis, and an accelerated erythrocyte sedimentation rate. X-ray imaging typically reveals extensive infiltrative bone destruction in the diaphysis of long bones or flat bones, presenting as a moth-eaten lytic pattern with poorly defined margins. Periosteal reaction is observed, often appearing as lamellated or "onion-skin-like" patterns.
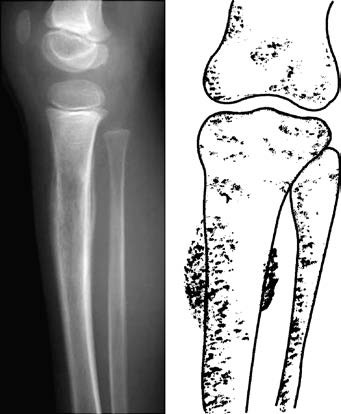
Figure 1 Ewing sarcoma in the upper segment of the tibia
Treatment
Ewing sarcoma is highly sensitive to radiotherapy. The tumor shrinks rapidly following low-dose radiation, and local pain is significantly alleviated. However, due to the tumor's tendency for early metastasis, radiotherapy alone yields poor long-term outcomes. Chemotherapy is also effective, although the overall prognosis remains poor. Currently, a combination of radiotherapy, chemotherapy, and surgical intervention (including limb-sparing surgeries or amputation) is used, with survival rates exceeding 50%.