Giant cell tumor (GCT) of bone is a borderline or behaviorally indeterminate tumor that can be classified into benign giant cell tumors and malignant giant cell tumors. The benign form is a locally aggressive tumor composed of sheets of ovoid mononuclear stromal cells evenly interspersed with large osteoclast-like multinucleated giant cells. The malignant form primarily manifests as a sarcoma originating from giant cells or develops secondarily via malignant transformation of an existing benign giant cell tumor. GCT of bone most commonly occurs between the ages of 20 and 40, with a slightly higher prevalence in females. It is frequently found in the metaphyseal-epiphyseal regions of long bones and the vertebral bodies, particularly in the distal femur and proximal tibia.
The tumor tissue consists primarily of mononuclear stromal cells and multinucleated giant cells. Based on the degree of differentiation and quantity of these two cell types, Jaffe classified giant cell tumors into three grades: Grade I is characterized by sparse stromal cells, few mitotic figures, and numerous multinucleated giant cells. Grade II shows dense stromal cellularity, a moderate number of mitotic figures, and a reduced population of multinucleated giant cells. Grade III is dominated by stromal cells, with marked nuclear atypia, abundant mitotic figures, and very few multinucleated giant cells. Accordingly, Grade I is regarded as benign, Grade II as intermediate, and Grade III as malignant. Although the biological behavior, radiological features, and benign/malignant nature of the tumor do not always correspond directly to the pathological grade, Jaffe’s classification provides valuable reference in determining tumor characteristics, severity, and the treatment plan.
Clinical Features
The primary symptoms include pain and swelling, which are related to the progression of the disease. A localized mass may present as having a "ping-pong ball" consistency to palpation, along with tenderness. Joint function near the lesion may be restricted. The typical X-ray findings include an eccentric, lytic, cystic lesion within the epiphysis without periosteal reaction. The lesion often demonstrates expansile growth, thinning of the cortical bone, and a "soap bubble"-like appearance. Highly aggressive tumors may breach the cortical bone, leading to pathological fractures. Angiographic imaging often reveals a hypervascular tumor, sometimes with arteriovenous fistula formation.
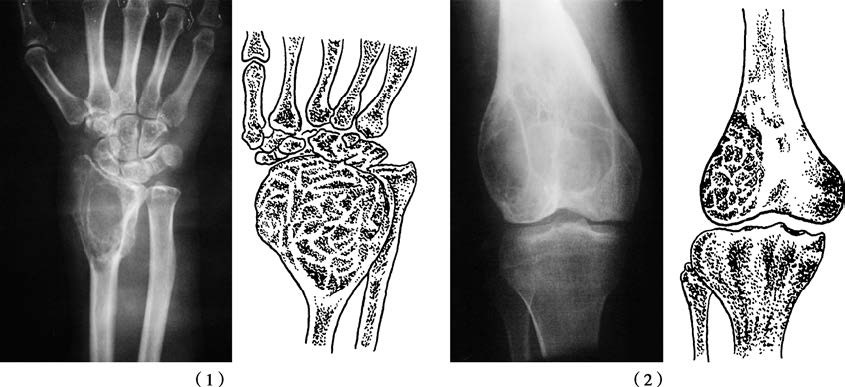
Figure 1 Giant cell tumor of bone
(1) Giant cell tumor of the distal radius
(2) Giant cell tumor of the distal femur
Treatment
For cases classified as G0T0M0–1), surgical resection is the primary treatment. This involves resection with adjuvant inactivation techniques, followed by reconstruction using autografts, allografts, or bone cement, although the risk of recurrence remains high. Recurrence cases require further intervention, including complete resection, segmental resection, or prosthesis implantation. In patients classified as G1–2T1–2M0, wide or radical resection is performed, as chemotherapy has limited efficacy. For tumors in surgically challenging locations, such as the spine, radiotherapy and chemotherapy may be considered. However, radiotherapy increases the risk of sarcomatous transformation and requires careful monitoring.
Denosumab, a specific inhibitor of receptor activator of nuclear factor kappa-B ligand (RANKL), has shown promise in the treatment of unresectable or refractory giant cell tumors, aiding in disease control and reducing recurrence.