Etiology and Pathology
The etiology of pheochromocytoma and paraganglioma (PPGL) remains unclear. Research indicates that approximately 30% of cases have a hereditary background and may be associated with genetic syndromes such as von Hippel-Lindau disease (VHL disease), multiple endocrine neoplasia type 2 (MEN-2), familial paraganglioma types 1–5, and neurofibromatosis type I. These conditions have been linked to gene mutations affecting pseudohypoxia pathways and kinase pathways. Around 39% of sporadic PPGL cases exhibit pathogenic genetic mutations, with some tumors also showing epigenetic alterations such as hypermethylation and changes in microRNAs (miRNAs).
Pheochromocytomas (PHEO) are typically unilateral, although hereditary cases often present as bilateral and multifocal lesions. Most tumors are large, with variable morphology, appearing as spherical or lobulated masses with a well-defined capsule. On sectioning, lesions appear reddish-brown, highly vascularized, and may show areas of hemorrhage, necrosis, and cystic degeneration. Tumor cells exhibit abundant cytoplasm, which may appear granular, filamentous, or vacuolated. Yellow-brown granules indicative of chromaffin positivity can be observed in the cytoplasm of tissue fixed with dichromate. The etiology of adrenal medullary hyperplasia remains uncertain; it may occur as an isolated condition or as bilateral lesions with varying degrees of hyperplasia. Its clinical manifestations are similar to those of PPGL.
Clinical Manifestations
Catecholamine hypersecretion is most common in individuals aged 30–50 years and presents with diverse symptoms primarily caused by elevated catecholamines in the blood. Major symptoms include hypertension and metabolic disturbances.
Headache, Palpitations, and Sweating
These are the classic triad of symptoms for pheochromocytoma, demonstrating over 90% specificity and sensitivity for the diagnosis of PPGL.
Hypertension
Symptoms include paroxysmal hypertension superimposed on baseline hypertension. During episodes, patients may experience severe headaches, pallor or flushing, cold extremities, nausea, vomiting, profuse sweating, palpitations, shortness of breath, and visual disturbances. Over 40% of cases involve paroxysmal hypertension, which is more common in females. Some patients may appear normotensive under normal conditions but exhibit sudden blood pressure spikes in response to external stimuli, such as emotional stress, trauma, pregnancy, childbirth, anesthesia, or surgery.
Metabolic Disturbances
Excess catecholamine secretion disrupts various metabolic processes. Common effects include increased basal metabolic rate, accelerated glycogenolysis, suppressed insulin secretion leading to elevated blood glucose and glycosuria, and enhanced fat metabolism, resulting in elevated serum free fatty acid and cholesterol levels. A minority of patients may present with hypokalemia.
Catecholamine-Induced Cardiomyopathy
This is a severe and distinctive complication of PHEO. Persistent or intermittent release of large amounts of catecholamines into the bloodstream can cause myocardial cell swelling, degeneration, and focal necrosis, eventually leading to myocardial fibrosis. Clinical manifestations include arrhythmias, heart failure, myocardial hypertrophy, and myocardial ischemia.
Skin Symptoms
Catecholamine-induced vasoconstriction results in pallor and reduced skin temperature, particularly in the extremities.
Auxiliary Examinations
Laboratory Tests
24-Hour Urinary Catecholamine Measurement
This includes adrenaline, noradrenaline, and dopamine. A catecholamine level exceeding twice the normal upper limit in 24-hour urine samples is considered significant.
Plasma Free Metanephrines (MNs) Measurement
This includes metanephrine (MN) and normetanephrine (NMN), and is suitable for screening and monitoring high-risk individuals.
24-Hour Urinary Vanillylmandelic Acid (VMA) Measurement
VMA is a metabolite of adrenaline and noradrenaline and is primarily excreted in the urine. Certain dietary items and medications (e.g., coffee, bananas, citrus fruits, aspirin) can interfere with test values and should be discontinued prior to testing.
Localization Studies
Ultrasound (US)
This is commonly used for screening purposes. Detection rates for adrenal pheochromocytomas are high, especially for tumors larger than 3 cm in diameter.
CT Scanning
Pheochromocytomas are detectable with nearly 100% accuracy. Tumors usually exhibit inhomogeneous density and marked contrast enhancement. CT scans also help assess the relationship of the tumor to surrounding blood vessels and organs.
MRI Scanning
MRI has a sensitivity of 95% and a specificity of 100% in diagnosing pheochromocytomas. It is an optimal imaging modality for children, pregnant women, patients requiring reduced radiation exposure, or those allergic to CT contrast agents.
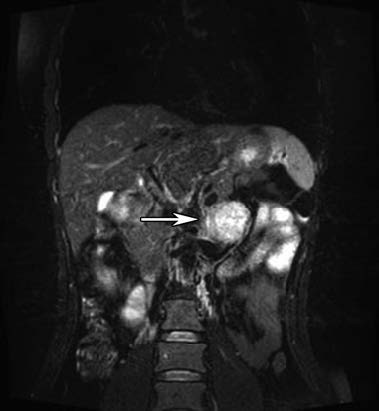
Figure 1 MRI of left adrenal pheochromocytoma
131I-Metaiodobenzylguanidine (131I-MIBG) Scintigraphy
This compound is absorbed by chromaffin cells, and radiolabeled isotopes allow for the identification of pheochromocytoma location.
Somatostatin Receptor Imaging
Somatostatin receptors are G protein-coupled transmembrane proteins with five subtypes, primarily type 2 and type 4 expressed in PPGL. Octreotide, a somatostatin analog, is radiolabeled for somatostatin receptor scintigraphy, aiding in pheochromocytoma localization.
PET Imaging
Modalities such as 18F-FDG PET, 11C-hydroxyephedrine PET, 11C-epinephrine PET, and 18F-DOPA PET (dopaminergic imaging) have been reported for PPGL localization with high sensitivity and specificity.
Diagnosis
The diagnosis of catecholamine excess is primarily based on significant blood pressure elevation, particularly in cases of malignant or paroxysmal hypertension. Adrenal tumors demonstrating inhomogeneous enhancement on CT or high signal intensity on T2-weighted MRI are suggestive findings. Positive results from blood or urinary catecholamine measurements further support the diagnosis of pheochromocytoma. Occasionally, even in the absence of abnormalities in laboratory tests, pheochromocytoma remains a clinical consideration.
Treatment
Surgical Treatment
Laparoscopic, robot-assisted, or open resection of the tumor yields favorable outcomes. Due to prolonged catecholamine-induced vasoconstriction in patients with pheochromocytoma, relative hypovolemia may occur. Following tumor resection, the sudden reduction of catecholamine levels can lead to vasodilation, potentially causing intraoperative or postoperative hypovolemic shock, which can be life-threatening. Comprehensive preoperative preparation, meticulous intraoperative management, and close postoperative monitoring are essential to prevent complications.
For bilateral, familial, or genetically predisposed cases, preservation of normal adrenal tissue is recommended to avoid lifelong glucocorticoid replacement.
Medical Treatment
For patients unable to undergo surgery, those with unresected pheochromocytomas, or in the case of tumor recurrence following surgery, medications such as alpha-adrenergic receptor antagonists may be used to alleviate symptoms. Internal radiotherapy with 131I-MIBG is also a therapeutic option.